
The MIFETM
system
for non-invasive measurement of specific fluxes in solution
near living
plant or animal tissue

|
|
The MIFETM
system
| |
|
Home UTas Biophysics Lab MIFE user group Overview MIFE theory Hardware Software Purchasing |
Principles and basic theory of microelectrode ion flux measurementChemicals in solution move under the influence of chemical forces of diffusion directed towards lower concentration regions. Ions, which are charged, also experience electrical forces if an electric field is present as well. The movement of an ion in solution can be described in terms of these chemical and electrical driving forces and other parameters of the ion and solution. It can be shown that the net flux of an ion, typically measured in units of nmol m-2 s-1, may be found from a measurement of the change in voltage of an ion selective microelectrode that is moved through a small known distance in the solution. This technique allows non-invasive measurement of net ion fluxes through a tissue boundary with resolution of 10 seconds in time and 20 micrometer in position. A suitable microscope is used to observe the microelectrodes and the tissue near which they are moved.In the diagram a
microelectrode, whose tip is filled with the liquid
ion exchanger LIX, is initially at a distance x from the tissue into
which
ions are moving with a net flux J. Basic electrochemical
theory (See Newman, 2001) shows that the net ionic flux J
is given in terms of the ion concentration c (mol m-3),
the mobility of the ion u (speed per unit force, m s-1
per newton mol-1), and the force per mole which
is the electrochemical
potential gradient (dµ/dx). Thus J
= c u
(dµ/dx). But dµ
is the same inside the
electrode as in the bath solution, and in the electrode dµ
= zFdV because the concentration inside is fixed.
Hence the flux
may be written J = c u z F(dV/dx).
The concentration
is known, or is adequately measured by the value of V
when the electrode
has been calibrated in standard solutions. For the ion, u
and z
are known constants, although for multi-valent
ions u depends on z.
The electrometer measures dV as the electrode
is moved through the chosen distance dx. In practice, the LIX is not ideal, so the electrode is calibrated to find its actual "Nernst slope" (which depends on the valence). The theory can also be
expressed in terms of the the diffusion coefficient
D
( = u R T) for the ion instead of the mobility u. The fluxes of neutral molecules, eg O2 and CO2, can also be measured using the technique. An example is Pang et al (2006).
|
Maintained by Ian Newman. Date . © University of Tasman
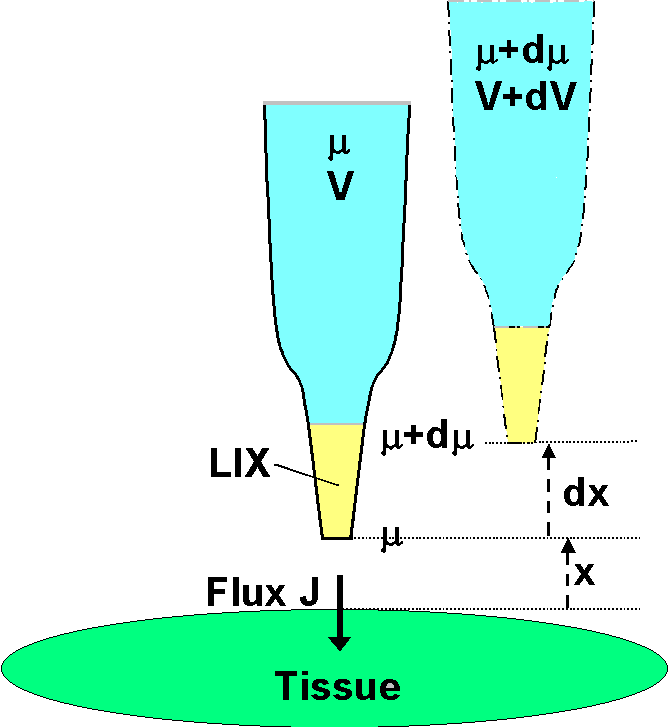 It
is assumed that there is no bulk solution flow, so that ionic movement
in the solution (regardless of the membrane transport processes) is
solely
by diffusion under the influence of electric and chemical forces in
solution.
It is also assumed that the measurement is close to the surface and that
the
ionic movement is normal to the surface. The electrochemical potential
in the solution at the distance x is µ (joules mol-1).
Because the LIX allows free passage of the ion in question (but no
others),
the electrochemical potential of the ion inside the electrode is also
µ.
The chemical component of µ inside the electrode is fixed by the
concentration of the filling solution; the electrical component is
given
by zFV , where V (volts) is
measured by an electrometer connected
via suitable half cells to the electrode solution and to a reference
electrode
some distance away in the bath solution. The ion's valence is
z
and F is the Faraday number. The microelectrode is
now moved slowly away
(not to disturb the solution) through a small distance
dx.
(It is shown offset in the diagram for clarity only.) At this new
position
in solution the electrochemical potential is µ + dµ.
It is the same inside the electrode, but only the electrical component
there has changed, and the measured voltage is now V
+ dV.
It
is assumed that there is no bulk solution flow, so that ionic movement
in the solution (regardless of the membrane transport processes) is
solely
by diffusion under the influence of electric and chemical forces in
solution.
It is also assumed that the measurement is close to the surface and that
the
ionic movement is normal to the surface. The electrochemical potential
in the solution at the distance x is µ (joules mol-1).
Because the LIX allows free passage of the ion in question (but no
others),
the electrochemical potential of the ion inside the electrode is also
µ.
The chemical component of µ inside the electrode is fixed by the
concentration of the filling solution; the electrical component is
given
by zFV , where V (volts) is
measured by an electrometer connected
via suitable half cells to the electrode solution and to a reference
electrode
some distance away in the bath solution. The ion's valence is
z
and F is the Faraday number. The microelectrode is
now moved slowly away
(not to disturb the solution) through a small distance
dx.
(It is shown offset in the diagram for clarity only.) At this new
position
in solution the electrochemical potential is µ + dµ.
It is the same inside the electrode, but only the electrical component
there has changed, and the measured voltage is now V
+ dV.